The Changing Landscape of a Terrifying Diagnosis
What Exactly Happens inside the Pulmonary Arteries?
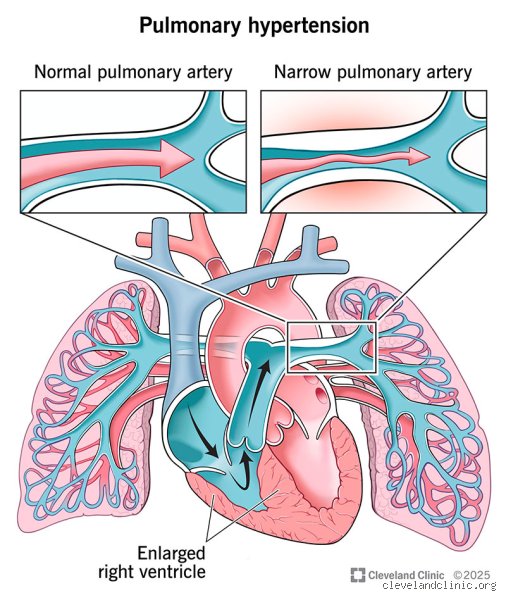
To understand why people used to view this disease as uniformly fatal, you have to look at the sheer physics of the heart. Pulmonary arterial hypertension isn't your run-of-the-mill high blood pressure that you check with an arm cuff at a pharmacy. Instead, the condition targets the microscopic arteries in the lungs, causing them to narrow, stiffen, and become scarred. The right ventricle of the heart, which is a thin-walled muscle designed to pump blood under low pressure into the lungs, suddenly finds itself pushing against a brick wall. Imagine trying to force thick fluid through a tiny straw—eventually, the pump fails. This specific form of right-sided heart failure, known medically as cor pulmonale, remains the primary cause of mortality in patients. Yet, the architectural remodeling of these vessels happens silently, often masking itself as simple fatigue or out-of-shape breathlessness during everyday activities like climbing stairs.
The Grim Shadows of Historical Statistics
Honestly, it's unclear why some medical practitioners still quote data from the late 1980s, but doing so does a massive disservice to patients. In 1987, the National Institutes of Health (NIH) established the first major registry for the disease, revealing a devastating median survival time of just 2.8 years from diagnosis without targeted treatment. That specific number is what haunts the internet. But we are far from that era now. Because back then, doctors had virtually nothing in their toolkit besides lung transplantation, blood thinners, and oxygen tanks. It was a bleak landscape where a diagnosis of idiopathic PAH felt identical to an advanced stage cancer prognosis, which explains why the terrifying reputation of the disease persists today despite radical shifts in cardiology and pulmonology.
Decoding the Mechanics: Why Medical Consensus Disagrees on Exact Outcomes
The Illusion of a Single Disease Entity
Where it gets tricky is that PAH isn't a monolith. The World Health Organization classifies pulmonary hypertension into five distinct groups, and PAH specifically represents Group 1. Within this single group, outcomes diverge wildly depending on the underlying trigger. For instance, a patient diagnosed with idiopathic PAH—meaning it arises spontaneously without a known cause—often carries a different risk profile compared to someone whose condition is secondary to systemic sclerosis or scleroderma. Why does this matter? Well, autoimmune-associated PAH tends to be significantly more aggressive, frequently resisting standard vasodilator therapies. On the flip side, patients who develop the condition due to a congenital heart defect that was repaired later in life might experience a much slower, more indolent disease progression. Experts disagree on whether we should even group these patients under the same prognostic umbrella, given that their cellular pathways differ so dramatically.
The Real-World Numbers in the 2020s
Let's look at actual data rather than historical panic. Modern registries, such as the REVEAL registry in the United States and the COMPERA network across Europe, paint a completely different picture of life expectancy. Today, the one-year survival rate sits at roughly 85% to 90%, while the five-year survival rate hovers around 60% to 65% across broad patient cohorts. More importantly, for individuals categorized as "low risk" after their initial treatment phase, the survival numbers climb even higher. I believe we must stop treating the disease as a ticking clock and start viewing it through the lens of risk stratification. It is a shifting scale. A person's trajectory depends entirely on hemodynamics measured during a right heart catheterization, such as a pulmonary vascular resistance of less than 5 Wood units or a right atrial pressure below 8 mmHg, which signal a far more optimistic future.
The Triad of Pathways: How Science Halts Vascular Remodeling
Targeting the Nitric Oxide, Endothelin, and Prostacyclin Systems
The transition away from an invariably fatal outcome occurred because scientists mapped out three specific chemical pathways that control blood vessel constriction. First, the prostacyclin pathway, which is often deficient in sick lungs. By introducing synthetic prostacyclins—such as epoprostenol, which requires a continuous intravenous infusion via a pump that patients wear 24 hours a day—we can forcibly dilate these stubborn vessels. Then, there is the endothelin pathway. Endothelin-1 is a potent vasoconstrictor that exists in excess in these patients, so we deploy oral endothelin receptor antagonists like bosentan or macitentan to block its action. Finally, therapies targeting the nitric oxide pathway, including phosphodiesterase-5 inhibitors, help relax the smooth muscle cells. The true revolution happened when clinicians realized that hitting just one pathway wasn't enough; instead, using initial oral combination therapy right out of the gate is what truly alters the survival curve.
The Game-Changer: Sotatercept and Activin Signaling
People don't think about this enough, but for decades, all our medications merely dilated vessels without fixing the underlying cellular cellular proliferation. That changed recently with the FDA approval of sotatercept in 2024, a first-in-class biologic that targets activin signaling to actually reverse the vascular remodeling. It acts like a molecular brake on the runaway cell growth inside the arterial walls. In clinical trials, patients using this drug showed a dramatic reduction in pulmonary artery pressures and a significant improvement in their six-minute walk distance. It represents a paradigm shift from merely managing symptoms to potentially repairing the structural damage within the pulmonary circulation system itself.
Evaluating Risk: Phenotypes and the Varied Trajectories of Patients
Why Age and Comorbidities Dictate the Survival Timeline
But here is the catch: a twenty-something college student diagnosed with heritable PAH due to a BMPR2 gene mutation will face a completely different reality than a seventy-year-old lifetime smoker with underlying metabolic syndrome. The younger patient has a vascular system that is highly resilient, meaning they can tolerate aggressive triple-combination therapy—even continuous intravenous drugs that require meticulous central line care—allowing them to live for decades or bridge safely to a bilateral lung transplant. Conversely, an elderly patient often presents with what cardiologists call a "left heart phenotype," where stiff myocardial tissue complicates the treatment of lung pressures. If you pump vasodilators into a system where the left side of the heart cannot handle the increased blood flow, you risk causing pulmonary edema, a dangerous fluid backup in the lungs. Hence, age and cardiac compliance remain the ultimate gatekeepers of longevity in this medical domain.
Common mistakes and misconceptions about pulmonary arterial hypertension
People often conflate pulmonary hypertension with basic high blood pressure. They are entirely separate beasts. Systemic hypertension means your arm cuff reads high, whereas pulmonary arterial hypertension targets the restricted, high-resistance arena of your pulmonary vasculature. Another frequent blunder is treating the condition as a immediate, unavoidable death sentence. Is PAH always fatal? Historically, the grim timeline gave patients a mere 2.8 years of survival post-diagnosis. That was the baseline in the 1980s. Today, treating this illness with outdated statistics is an absolute disservice to modern science. The problem is that many internet resources still parrot these ancient, terrifying survival rates as if they were gospel.
The myth of the uniform progression
Many assume every patient follows a identical, rapid downward spiral. This is patently false. Clinical phenotypes vary wildly between an individual with idiopathic disease and someone whose condition stems from scleroderma. Why do we pretend one prognosis fits all? Because it simplifies medical jargon, except that simplification kills hope. Some individuals remain stable on oral dual therapy for over a decade. Others require aggressive intravenous prostanoids within months. Assuming a linear decline prevents physicians from tailoring therapies to the specific kinetic profile of the patient's remodeling vessels.
Misinterpreting idiopathic labels as hopeless
When a specialist diagnoses idiopathic PAH, patients frequently hear "untreatable" instead of "unknown origin". Let's be clear: an unknown trigger does not imply a lack of therapeutic targets. Modern medicine successfully manipulates the nitric oxide, endothelin, and prostacyclin pathways regardless of what initiated the cellular malfunction. Falling into the trap of assuming an idiopathic diagnosis guarantees a fatal outcome ignores thirty years of pharmacological evolution. It stops people from seeking specialized centers where advanced care actually exists.
The hidden paradigm: Right ventricular adaptation and expert intervention
The true battleground of this disease is not actually the lungs. It is the right ventricle of the heart. As the pulmonary arteries stiffen and narrow, the right ventricle must pump against an escalating wall of resistance. Experts do not just look at lung pressures; they obsess over right ventricular function and remodeling. The heart possesses an astonishing, albeit finite, capacity to hypertrophy and adapt to this intense workload. Yet, this compensatory mechanism eventually tires out, which explains why monitoring right ventricular ejection fraction via cardiac MRI has become a gold standard for survival prediction.
The power of proactive, triple-combination therapy
Waiting for a patient to deteriorate before maximizing medication is a fatal strategy. Upfront triple-combination therapy—utilizing an endothelin receptor antagonist, a PDE5 inhibitor, and a prostacyclin analogue simultaneously—has shifted the entire treatment paradigm. Early aggressive intervention destroys the traditional prognosis by stopping vascular remodeling before the tissue undergoes irreversible fibrosis. Data indicates that hitting the disease hard during the early functional classes preserves right ventricular geometry. This proactive stance turns a rapidly progressive condition into a manageable, chronic vascular disease for a significant portion of the cohort.
Frequently Asked Questions
What is the current life expectancy for someone diagnosed with pulmonary arterial hypertension?
Recent registry data from the REVEAL study indicates that the overall five-year survival rate for patients has risen to approximately 65 percent, a monumental leap from previous decades. For individuals categorized as low-risk using comprehensive risk-stratification scores, the three-year survival probability exceeds 95 percent. Conversely, high-risk individuals face a more precarious timeline, often showing a one-year survival rate of roughly 70 percent if aggressive interventions are not deployed immediately. These numbers demonstrate that the question of whether is PAH always fatal has been answered with a resounding no, provided modern medical protocols are utilized. Geography and access to an accredited pulmonary vascular center remain major variables in these statistical outcomes.
Can lifestyle modifications reverse the damage caused by pulmonary arterial hypertension?
Dietary changes and exercise cannot repair the structural remodeling, endothelial dysfunction, or plexiform lesions present in advanced pulmonary arterial hypertension. Patients must understand that sodium restriction, typically limited to less than 2,000 milligrams per day, merely prevents fluid overload and eases the workload on a failing right ventricle. Supervised, low-intensity cardiopulmonary rehabilitation can improve a patient's six-minute walk distance by an average of 30 to 40 meters, reflecting better muscular oxygen extraction rather than a cure within the pulmonary bed itself. (Prostacyclin therapies remain the true heavy lifters for vascular restructuring). In short, lifestyle adjustments keep the body viable and prevent acute right-heart failure episodes, but they function strictly as a supportive scaffold alongside intense, lifelong pharmacological regimens.
Is a lung transplant the only definitive cure available for patients?
A double-lung or heart-lung transplant is not technically a cure, but rather a complex exchange of one life-limiting condition for another. Survival rates following transplantation hover around 50 to 60 percent at the ten-year mark, requiring a lifetime of toxic immunosuppressive drugs to prevent organ rejection. Physicians reserve this drastic surgical option exclusively for patients who have failed maximal medical therapy and remain in WHO Functional Class IV. Because of the limited availability of donor organs and the strict eligibility criteria, less than five percent of the global patient population undergoes this procedure annually. As a result: medication remains the primary long-term survival strategy for the vast majority of individuals fighting this condition.
A definitive look at the survival landscape
We must stop discussing this vascular disease using the fatalistic vocabulary of the twentieth century. Is PAH always fatal? Absolutely not, and continuing to frame it as an inevitably rapid death sentence is a destructive medical narrative that dampens patient compliance and hinders clinical optimism. The terrifying statistics found on random internet searches reflect historical cohorts who lacked access to modern, multi-pathway targeting. Our contemporary reality features patients living vibrant lives decades after their initial catheterization. Survival is no longer an anomaly; it is the target we aggressively chase with upfront, triple-combination therapies. The medical community must hold a firm line against defeatism, ensuring every newly diagnosed individual is immediately funneled to an expert center. We possess the tools to stall this disease, reshape the right ventricle, and radically extend human life.