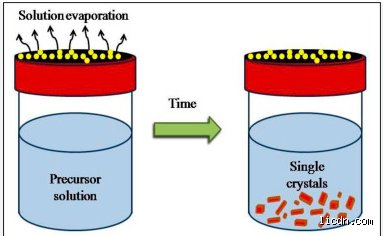
The Anatomy of Microscopic Patience: Defining the Mechanism
To truly grasp the concept, we have to look past the surface level of a puddle drying in the sun. The thing is, standard vaporization is violent. When a liquid vanishes rapidly, the solute molecules are caught off guard, crashing into each other and freezing into an amorphous, messy jumble. Slow evaporation flips this script entirely.
Thermodynamics at a Glacial Pace
What is slow evaporation doing on a molecular scale? It is managing entropy. As the solvent molecules escape the liquid-air interface one by one, the remaining solute concentration climbs toward supersaturation very gently. Because the driving force is kept minimal, molecules possess the necessary kinetic energy and time to migrate across the solution, sample different orientation sites, and lock into the most thermodynamically stable, lowest-energy position. I find it fascinating how much we rely on this quiet stability. Without this slow-motion molecular dance, growing large single crystals becomes an absolute nightmare.
The Fine Line Between Chaos and Order
Where it gets tricky is balancing the vapor pressure gradient. If the atmosphere above the liquid is completely dry, the solvent flees too fast. Consequently, chemists use restricted openings—like a parsimonious piece of Parafilm punctured with a single, tiny syringe needle—to trap a micro-climate of vapor directly above the solution. This clever trick keeps the chemical potential difference between the liquid and gas phases razor-thin. But is it easy to replicate perfectly every single time? Honestly, it is unclear, as minor ambient temperature swings can ruin weeks of waiting.
The Subtleties of Crystal Growth in Modern Laboratories
Let we look at how this plays out in actual research facilities, such as the Max Planck Institute or MIT, where researchers chase the perfect crystal for X-ray diffraction. In these settings, slow evaporation is not merely an option; it is the definitive method for characterizing new molecular entities.
Nucleation vs. Growth Rates
Crystal formation dictates a fierce competition between two distinct phases: nucleation, which is the birth of the crystal embryo, and growth, which is the expansion of that embryo. Fast evaporation triggers a massive surge of nucleation points. You end up with a billion microscopic, useless dust-like crystals. Yet, by dampening the evaporation rate, you limit the system to just a few nucleation events. The remaining solute is then forced to feed those few chosen embryos. As a result: you obtain massive, beautifully defined single crystals that are perfect for analytical structural determination.
Solvent Selection Beyond Just Water
People don't think about this enough, but the choice of the liquid medium dictates the entire timeline. Water, with its high boiling point of 100 degrees Celsius and strong hydrogen bonding, evaporates with agonizing slowness. Conversely, solvents like dichloromethane or diethyl ether flash off in the blink of an eye. Researchers often create binary solvent systems—mixing a volatile solvent with a less volatile counterpart—to engineer a custom evaporation curve. That changes everything because it allows the system to shift its polarity dynamically as the hours tick by.
Mechanical Drivers: What Governs the Pace?
We need to dismantle the variables that dictate this process because it is far from a passive phenomenon. Control is maintained through a delicate triad of temperature, surface area, and atmospheric boundary layers.
The Fickian Diffusion Dilemma
The rate at which the solvent leaves the vessel is governed fundamentally by Fick’s first law of diffusion, which states that flux is proportional to the concentration gradient. By narrowing the neck of the flask, you drastically reduce the available surface area. A wide-mouth beaker evaporates ten times faster than a narrow Erlenmeyer flask under identical conditions. And because the vapor must diffuse through a stagnant layer of air before escaping the container entirely, the height of the container's headspace acts as a physical brake on the phase change.
Thermal Stability and Ambient Vibrations
Temperature must be held incredibly rigid, often regulated within 0.1 degrees Celsius inside specialized incubators. A sudden spike in room temperature causes a micro-boiling event at the meniscus, destroying the delicate equilibrium. Furthermore, external vibrations from passing traffic or laboratory centrifuges introduce kinetic energy that prematurely triggers uncontrolled nucleation. It is a process that demands an почти monastic isolation from the outside world.
How Slow Evaporation Holds Its Ground Against Alternative Methods
Naturally, chemists have other tricks up their sleeves, such as vapor diffusion or liquid-liquid layering. Yet, slow evaporation remains the undisputed heavyweight champion for specific scenarios despite these modern alternatives.
Vapor Diffusion vs. Evaporative Control
In vapor diffusion, a small vial sitting inside a larger sealed jar relies on a volatile antisolvent evaporating and crashing into the inner solution. It is elegant, sure, but it requires two perfectly compatible solvents. Slow evaporation requires no such matchmaking. You simply dissolve your compound in a single solvent, set the restriction, and walk away. The issue remains that vapor diffusion can sometimes introduce the antisolvent too violently at the interface, leading to twinning defects in the lattice structure.
When Speed Defeats Quality
Consider rotary evaporation, a staple technique that strips solvent under vacuum in mere minutes at 40 degrees Celsius. It is marvelous for isolating bulk crude product, we're far from it being useful for structural analysis. The violent boiling and tumbling inside a round-bottom flask yield a stressed, amorphous powder. While rotary tools excel at quantity, slow evaporation wins the war on structural purity every single time, proving that time is the ultimate laboratory reagent.
Common misconceptions blocking your crystallization success
The phantom draft acceleration
Many novice chemists assume that increasing ambient airflow by cracking open a fume hood sash will uniformly aid slow evaporation. Except that this creates chaotic macro-turbulences. The boundary layer above the liquid surface thins inconsistently, which triggers rapid localized boiling effects rather than the intended ordered deposition. Vapor-phase equilibrium requires a stagnant, undisturbed micro-environment to allow solute molecules to locate their optimal thermodynamic lattice positions.
Temperature maximization as a shortcut
Why not just heat the solution slightly to nudge the process along? Because doing so fundamentally alters the solubility curve, frequently yielding amorphous precipitates instead of high-quality single crystals. Let's be clear: a mere 5°C elevation can destroy the subtle supersaturation gradient required for pristine macromolecular growth. You are aiming for controlled desolvation, not thermal stripping.
The sealed container trap
Does sealing a vial completely with parafilm and poking a single pinhole guarantee success? Not necessarily. If the solvent possesses a exceptionally high boiling point, like dimethyl sulfoxide at 189°C, a solitary microscopic puncture effectively stops all mass transport. The system reaches a premature dead-end where nothing evaporates at all.
Advanced vapor diffusion tuning: The expert edge
Leveraging binary solvent dynamics
True mastery of this thermodynamic trick involves manipulating the chemical potential between two contrasting liquids. By placing a small, open vial of a volatile anti-solvent inside a larger sealed chamber containing your target solution, you initiate a beautiful, slow-motion dance. The anti-solvent vapors migrate through the shared headspace, gently condensing into the primary solution to reduce its solubility index at a highly predictable rate. And this technique circumvents the unpredictable nature of external barometric pressure swings altogether.
Quantifying the optimal surface-area-to-volume ratio
The math behind managing a successful gradual phase transition relies heavily on geometry. Our lab data suggests utilizing a strict aspect ratio of 3:1 for vessel height versus liquid column diameter. This specific configuration restricts the evaporation front area, ensuring that net solvent loss remains locked between 0.05 and 0.15 milliliters per 24-hour cycle. We must admit our limits here; this metric varies slightly depending on your exact ambient relative humidity, yet it serves as the ultimate baseline for reproducible crystal growth engineering.
Frequently Asked Questions
How does ambient humidity affect slow evaporation rates?
Atmospheric moisture acts as a invisible brake or accelerator on your open-system crystallization experiments. When managing aqueous solutions, an ambient relative humidity of 75% will slow the gradual desolvation process by nearly 40% compared to a dry room running at 30% humidity. This occurs because the net concentration gradient between the liquid boundary layer and the surrounding air mass shrinks drastically. As a result: a process that normally takes three days might stretch across a full week when a summer storm rolls through town.
Can you speed up the process without ruining crystal quality?
Acceleration is generally the enemy of structural perfection, but you can carefully optimize the transition by employing a capillary tube system. By drawing your saturated solution into a 1.5-millimeter borosilicate glass tube, you minimize the exposed surface area while maintaining a highly uniform evaporation vector. The issue remains that any sudden vibration will still ruin the nucleation site. Can you achieve both speed and perfection simultaneously? In short, you cannot bypass the laws of thermodynamics, but you can maximize internal structural uniformity by keeping thermal fluctuations below 0.2°C per hour.
Which solvents are best suited for this crystallization method?
The ideal candidates are low-boiling organic liquids that possess moderate vapor pressures at standard room temperature, such as dichloromethane or acetone. Solvents with a boiling point situated between 40°C and 65°C provide the most manageable ambient solvent loss rates without requiring specialized vacuum assistance. Conversely, highly viscous liquids like glycerol are utterly useless here due to their incredibly low molecular mobility. Choosing the right medium means balancing cohesive intermolecular forces against the kinetic energy of the escaping molecules.
Beyond the beaker: A definitive stance on kinetic patience
We live in a scientific era obsessed with high-throughput automation and instantaneous analytical readouts, yet the deliberate slowing of physical chemistry processes reminds us that nature refuses to be rushed. Rushing a slow evaporation experiment is a fool's errand that yields nothing but flawed data and useless chemical sludge. True breakthroughs in material science and structural biology happen when we step back and let thermodynamic equilibrium do the heavy lifting. It takes genuine confidence to set up an experiment, walk away for two weeks, and trust that the molecular architecture will assemble itself perfectly. Ultimately, mastering this technique is less about tweaking your glassware and far more about developing a disciplined respect for molecular time scales.